
Síndrome de Borderline
O que é:
Síndrome de Borderline, ou transtorno de personalidade Limítrofe, é uma grave doença psicológica. Os indivíduos com a síndrome de Borderline vivem no limite entre a normalidade e os surtos psicóticos.
 As dificuldades para se relacionar, oscilações de humor e impulsividade são alguns dos problemas enfrentados pelo portador da síndrome de Borderline e pelas pessoas que o cercam. Esses sintomas começam a se manifestar na adolescência e se tornam concretos no inicio da vida adulta.
A síndrome de Borderline é frequentemente confundida com esquizofrenia ou transtorno bipolar, porém possui características diferentes, como a duração e intensidade das emoções....
As dificuldades para se relacionar, oscilações de humor e impulsividade são alguns dos problemas enfrentados pelo portador da síndrome de Borderline e pelas pessoas que o cercam. Esses sintomas começam a se manifestar na adolescência e se tornam concretos no inicio da vida adulta.
A síndrome de Borderline é frequentemente confundida com esquizofrenia ou transtorno bipolar, porém possui características diferentes, como a duração e intensidade das emoções....
Sintomas da Síndrome de Borderline
Oscilações de humor, agressividade, irritabilidade, depressão, automutilação, comportamentos suicidas, medo de abandono, dificuldade em lidar com as emoções, mudanças de planos profissionais e nos círculos de amizades, impulsividade e baixa autoestima.
Diagnóstico da Síndrome de Borderline
O diagnóstico é feito através de características do transtorno, observadas por um psicólogo ou psiquiatra, e por experiências relatadas pelo individuo. É importante fazer exames fisiológicos, como hemograma e sorologia, para a exclusão de outras doenças.
O diagnóstico da síndrome de Borderline pode ser longo e complexo. Devido à semelhança com outras síndromes, é muito raro obter o diagnóstico precoce dessa doença.
Tratamento da Síndrome de Borderline
O tratamento é feito através de uma combinação de medicamentos e de acompanhamento psicológico. Os medicamentos usados com mais frequência no tratamento da síndrome de Borderline incluem anti-depressivos, estabilizadores de humor e tranquilizantes.
A psicoterapia é o principal tratamento utilizado, porém requer paciência e força de vontade do paciente. É na psicoterapia que o individuo aprende a lidar e controlar as suas emoções
Síndrome de Pendred
 A síndrome de pendred é uma doença hormonal de origem genética caracterizada pela presença de bócio, uma alteraçao grave da tireóide, surdez profunda e mudez.
A doença é comum em indivíduos da mesma família e pode ser identificada através do aconselhamento genético. Um exame que avalia os cromossomos do indivíduo e vê a possiblidade do densenvolvimento de algumas doenças, deixando-o mais preparado psicológicamente para enfrentá-la.
Os portadores da doença devem tratar o bácio com medicamentos, mas não há cura para a sua surdez, sendo necessário aprender a língua gestual para comunicar-se.
Apesar desta limitação, o indivíduo portador da síndrome de pendred pode levar uma vida normal..
A síndrome de pendred é uma doença hormonal de origem genética caracterizada pela presença de bócio, uma alteraçao grave da tireóide, surdez profunda e mudez.
A doença é comum em indivíduos da mesma família e pode ser identificada através do aconselhamento genético. Um exame que avalia os cromossomos do indivíduo e vê a possiblidade do densenvolvimento de algumas doenças, deixando-o mais preparado psicológicamente para enfrentá-la.
Os portadores da doença devem tratar o bácio com medicamentos, mas não há cura para a sua surdez, sendo necessário aprender a língua gestual para comunicar-se.
Apesar desta limitação, o indivíduo portador da síndrome de pendred pode levar uma vida normal..
Síndrome do X Frágil
O que é:
A síndrome do X frágil é uma doença genética causada pela mutação de um gene no cromossomo X que geralmente afeta mais os meninos que as meninas.
Sintomas da síndrome do X frágil
São sintomas característicos da síndrome do X frágil:

- dificuldade de aprendizagem;
- face alongada;
- testículos grandes;
- orelhas e queixo salientes;
- baixo tônus muscular;
- hipotonia muscular;
- comprometimento do tecido conjuntivo;
- pés planos;
- palato alto;
- prolapso da válvula mitral;
- prega palmar única;
- estrabismo;
- escoliose;
- hábito de morder as mãos;
Algumas dessas características podem passar despercebidas até o inicio da adolescência, mas se tornam evidentes a partir dessa idade.
Diagnóstico da síndrome do X frágil
O diagnostico da síndrome do X frágil pode ser feito através de técnicas de biologia molecular, amostras de sangue, fio de cabelo ou líquido amniótico.
Tratamento para síndrome do X Frágil
O tratamento para síndrome do X Frágil é feito através de terapia comportamental, fisioterapia e, se for necessário, cirurgia para as anomalias físicas.
Os indivíduos com histórico de síndrome do X frágil na família devem procurar aconselhamento genético, para conhecer a probabilidade de ter filhos com a doença. Os homens tem o cariótipo XY, e se forem afetados podem transmiti-lo apenas para suas filhas, nunca para seus filhos uma vez que o gene recebido pelos meninos é o Y, e esse não apresenta nenhuma alteração relacionada com a doença.
Síndrome de Savant
 A síndrome de Savant é um distúrbio psíquico onde o indivíduo portador possui graves défices intelectuais. O portador desta síndrome tem dificuldades em se comunicar, compreender o que lhe é transmitido e estabelecer relações interpessoais. Mas, por outro lado, possui inúmeros talentos, principalmente ligados a uma extraordinária memória.
Alguns dos principais sintomas da síndrome de Savant são dificuldades para se relacionar com outras pessoas, resistência aos métodos de ensino, dificuldade em expressar as suas necessidades e habilidades motoras irregulares que aparecem ainda na infância. A síndrome tem maior incidência nos meninos.
Existem três tipos de Savant: os habilidosos, talentosos e os prodígios, que possuem facilidades em lidar com arte, cálculo e mecânica. A doença não é diagnosticada facilmente e pode ser confundida com o autismo. Mas a partir da observação das características do indivíduo é possível fazer o diagnóstico correto.
As causas da doença são desconhecidas e não existe cura para essa síndrome. Seu tratamento é direcionado para amenizar os sintomas, com o uso de terapia ocupacional, terapia da fala e hipoterapia, que proporcionam uma melhor qualidade de vida para os portadores da síndrome e para sua família.
A síndrome de Savant é um distúrbio psíquico onde o indivíduo portador possui graves défices intelectuais. O portador desta síndrome tem dificuldades em se comunicar, compreender o que lhe é transmitido e estabelecer relações interpessoais. Mas, por outro lado, possui inúmeros talentos, principalmente ligados a uma extraordinária memória.
Alguns dos principais sintomas da síndrome de Savant são dificuldades para se relacionar com outras pessoas, resistência aos métodos de ensino, dificuldade em expressar as suas necessidades e habilidades motoras irregulares que aparecem ainda na infância. A síndrome tem maior incidência nos meninos.
Existem três tipos de Savant: os habilidosos, talentosos e os prodígios, que possuem facilidades em lidar com arte, cálculo e mecânica. A doença não é diagnosticada facilmente e pode ser confundida com o autismo. Mas a partir da observação das características do indivíduo é possível fazer o diagnóstico correto.
As causas da doença são desconhecidas e não existe cura para essa síndrome. Seu tratamento é direcionado para amenizar os sintomas, com o uso de terapia ocupacional, terapia da fala e hipoterapia, que proporcionam uma melhor qualidade de vida para os portadores da síndrome e para sua família.
Síndrome Autista
 A síndrome autista manifesta-se na infância e é incurável. Medicamentos e a terapia comportamental são métodos que ajudam a lidar com a doença, diminuindo os seus sintomas e facilitando a vida do portador e de seus cuidadores.
Esta síndrome perdura por toda a vida e o portador terá grandes dificuldades nos relacionamentos interpessoais, o que pode vir a ter consequências no âmbito trabalhista.
Nem todos os autistas possuem défict de atenção e retardo mental, alguns são muito inteligentes e podem formar-se até na Universidade. Outros possuem maiores dificuldades necessitando de mais tempo para aprender, mas isso não quer dizer que ele não seja capaz de entender o mundo à sua volta.
Os pais de um portador da síndrome autista devem dedicar-se ao máximo ao seu filho. Apesar de não demonstrarem afeto, os autistas precisam de amor e carinho como qualquer outro indivíduo.
A síndrome autista manifesta-se na infância e é incurável. Medicamentos e a terapia comportamental são métodos que ajudam a lidar com a doença, diminuindo os seus sintomas e facilitando a vida do portador e de seus cuidadores.
Esta síndrome perdura por toda a vida e o portador terá grandes dificuldades nos relacionamentos interpessoais, o que pode vir a ter consequências no âmbito trabalhista.
Nem todos os autistas possuem défict de atenção e retardo mental, alguns são muito inteligentes e podem formar-se até na Universidade. Outros possuem maiores dificuldades necessitando de mais tempo para aprender, mas isso não quer dizer que ele não seja capaz de entender o mundo à sua volta.
Os pais de um portador da síndrome autista devem dedicar-se ao máximo ao seu filho. Apesar de não demonstrarem afeto, os autistas precisam de amor e carinho como qualquer outro indivíduo.
Síndrome da Vida Ocupada
 A Síndrome da Vida Ocupada é um distúrbio que afeta pessoas muito ocupadas e que têm uma vida estressante. As causas da Síndrome da Vida Ocupada são a vida agitada, sobrecarga de informação, insatisfação no trabalho e privação do sono.
A Síndrome da Vida Ocupada é um distúrbio que afeta pessoas muito ocupadas e que têm uma vida estressante. As causas da Síndrome da Vida Ocupada são a vida agitada, sobrecarga de informação, insatisfação no trabalho e privação do sono.
Sintomas da Síndrome da Vida Ocupada
Os sintomas da Síndrome da Vida Ocupada são:
- Pequenas falhas de memória frequentes que podem ser, por exemplo, não saber porque pegou ou onde deixou um determinado objeto, porque entrou em determinado local ou porque vai para um certo local;
- Dificuldade de concentração.
O individuo começa a sentir-se emocionalmente afetado e pode ter dificuldade em lidar com o stress provocado pela perda de memória, piorando o distúrbio. Muitas vezes a Síndrome da Vida Ocupada pode ser confundida com Doença de Alzheimer.
Tratamento da Síndrome da Vida Ocupada
O tratamento da Síndrome da Vida Ocupada não está estabelecido, mas existem alguns conselhos que poderão ajudar no seu tratamento e cura:
- Reorganizar a vida familiar e profissional, estabelecendo prioridades, não sobrecarregando a mente;
- Estabelecer tempo para descanso;
- Garantir a qualidade do sono com pelo menos sete horas de sono;
- Praticar exercício físico regularmente.
A toma de um remédio para a memória sob recomendação médica pode ser uma alternativa de tratamento para reduzir o principal sintoma da Síndrome da Vida Ocupada que é a perda de memória.
Síndrome de Asperger
 A Síndrome de Asperger é uma perturbação neurocomportamental de base genética, pode ser definida como uma perturbação do desenvolvimento que se manifesta por alterações sobretudo na interacção social na comunicação e no comportamento. Embora seja uma disfunção com origem num funcionamento cerebral particular, não existe marcador biológico, pelo que o diagnóstico se baseia num conjunto de critérios comportamentais.
A Síndrome de Asperger é uma perturbação neurocomportamental de base genética, pode ser definida como uma perturbação do desenvolvimento que se manifesta por alterações sobretudo na interacção social na comunicação e no comportamento. Embora seja uma disfunção com origem num funcionamento cerebral particular, não existe marcador biológico, pelo que o diagnóstico se baseia num conjunto de critérios comportamentais.
Entre as características mais comuns podemos destacar:
- Défice de comportamento social;
- Interesses limitados;
- Comportamentos rotineiros;
- Peculiaridade do discurso e da linguagem;
- Perturbação na comunicação não verbal;
- Descoordenação motora.
Como consequência destas dificuldades os portadores de Síndrome de Asperger acabam por se isolar e limitar os seus interesses a determinados temas assuntos, atitude que prejudica ainda mais a sua relação com o outro. Calcula-se que em Portugal existam cerca de 40.000 portadores de Síndrome de Asperger afectando maioritariamente os rapazes.
O Diagnóstico precoce é essencial para proporcionar aos portadores, os recursos necessários e a que têm direito que lhes permitam atingir o seu potencial, o qual muitas vezes é extraordinário, como pessoas verdadeiramente integradas na sociedade.
Acolher a Diferença e aceitá-la como um desafio é Missão de cada um de nós!
SINDROME DE DOWN
A síndrome de Down (SD) é uma alteração genética produzida pela presença de um cromossomo a mais, o par 21, por isso também conhecida como trissomia 21.
 A SD foi descrita em 1866 por John Langdon Down. Esta alteração genética afeta o desenvolvimento do individuo, determinando algumas características físicas e cognitivas. A maioria das pessoas com SD apresenta a denominada trissomia 21 simples, isto significa que um cromossomo extra está presente em todas as células do organismo, devido a um erro na separação dos cromossomos 21 em uma das células dos pais. Este fenômeno é conhecido como disfunção cromossômica. Existem outras formas de SD como, por exemplo: mosaico, quando a trissomia está presente somente em algumas células, e por translocação, quando o cromossomo 21 está unido a outro cromossomo.
O diagnóstico da SD se realiza mediante o estudo cromossômico (cariótipo), através do qual se detecta a presença de um cromossomo 21 a mais. Este tipo de análise foi utilizado pela primeira vez em 1958 por Jerome Lejeune.
Não se conhece com precisão os mecanismos da disfunção que causa a SD, mas está demonstrado cientificamente que acontece igualmente em qualquer raça, sem nenhuma relação com o nível cultural, social, ambiental, econômico, etc. Há uma maior probabilidade da presença de SD em relação à idade materna, e isto é mais freqüente a partir dos 35 anos, quando os riscos de se gestar um bebê com SD aumenta de forma progressiva.Paradoxalmente, o nascimento de crianças com SD é mais freqüente entre mulheres com menos de 35 anos, isto se deve ao fato de que mulheres mais jovens geram mais filhos e também pela influência do diagnóstico pré natal,que é oferecido sistematicamente às mulheres com mais de 35 anos.
Como a SD é uma alteração cromossômica, é possível realizar um diagnóstico pré natal utilizando diversos exames clínicos como, por exemplo, a amniocentese (pulsãotransabdominal do liquido amniótico entre as semanas 14 e 18 de gestação) ou a biópsia do vilo corial (coleta de um fragmento da placenta). Ambos os exames diagnosticam a SD e outrascromossopatias.
Recentemente a prática médica tem incorporado métodos para a determinação do risco de ter um filho com SD, como por exemplo, o exame bioquímico, que se realiza mediante a avaliação dos níveis de substâncias químicas no sangue materno alteradas no caso da SD. Este exame se realiza entre a semana 14 e 17. A ultrassonografia também pode colaborar para detectar a SD, através dos marcadores ecográficos, principalmente da prega nucal, que pode ser medida a partir da décima semana de gestação. Estas últimas intervenções não são consideradas diagnósticas, para isso é necessário realizar os exames mencionados em primeiro lugar.
Embora as alterações cromossômicas da SD sejam comuns a todas as pessoas, nem todas apresentam as mesmas características, nem os mesmos traços físicos, tampouco as malformações. A única característica comum a todas as pessoas é o déficit intelectual. Não existem graus de SD; a variação das características e personalidades entre uma pessoa e outra é a mesma que existe entre as pessoas que não tem SD.
Cerca de 50% das crianças com SD apresentam problemas cardíacos, algumas vezes graves, necessitando de cirurgia nos primeiros anos de vida.
A intervenção médica pode acontecer com a finalidade principal de prevenção dos problemas de saúde que podem aparecer com maior freqüência na SD. Queremos destacar que a SD não é uma doença e sim uma alteração genética, que pode gerar problemas médicos associados.
Devemos olhar a pessoas com SD em sua singularidade, para que possa ter um pleno desenvolvimento enquanto sujeito.
A SD foi descrita em 1866 por John Langdon Down. Esta alteração genética afeta o desenvolvimento do individuo, determinando algumas características físicas e cognitivas. A maioria das pessoas com SD apresenta a denominada trissomia 21 simples, isto significa que um cromossomo extra está presente em todas as células do organismo, devido a um erro na separação dos cromossomos 21 em uma das células dos pais. Este fenômeno é conhecido como disfunção cromossômica. Existem outras formas de SD como, por exemplo: mosaico, quando a trissomia está presente somente em algumas células, e por translocação, quando o cromossomo 21 está unido a outro cromossomo.
O diagnóstico da SD se realiza mediante o estudo cromossômico (cariótipo), através do qual se detecta a presença de um cromossomo 21 a mais. Este tipo de análise foi utilizado pela primeira vez em 1958 por Jerome Lejeune.
Não se conhece com precisão os mecanismos da disfunção que causa a SD, mas está demonstrado cientificamente que acontece igualmente em qualquer raça, sem nenhuma relação com o nível cultural, social, ambiental, econômico, etc. Há uma maior probabilidade da presença de SD em relação à idade materna, e isto é mais freqüente a partir dos 35 anos, quando os riscos de se gestar um bebê com SD aumenta de forma progressiva.Paradoxalmente, o nascimento de crianças com SD é mais freqüente entre mulheres com menos de 35 anos, isto se deve ao fato de que mulheres mais jovens geram mais filhos e também pela influência do diagnóstico pré natal,que é oferecido sistematicamente às mulheres com mais de 35 anos.
Como a SD é uma alteração cromossômica, é possível realizar um diagnóstico pré natal utilizando diversos exames clínicos como, por exemplo, a amniocentese (pulsãotransabdominal do liquido amniótico entre as semanas 14 e 18 de gestação) ou a biópsia do vilo corial (coleta de um fragmento da placenta). Ambos os exames diagnosticam a SD e outrascromossopatias.
Recentemente a prática médica tem incorporado métodos para a determinação do risco de ter um filho com SD, como por exemplo, o exame bioquímico, que se realiza mediante a avaliação dos níveis de substâncias químicas no sangue materno alteradas no caso da SD. Este exame se realiza entre a semana 14 e 17. A ultrassonografia também pode colaborar para detectar a SD, através dos marcadores ecográficos, principalmente da prega nucal, que pode ser medida a partir da décima semana de gestação. Estas últimas intervenções não são consideradas diagnósticas, para isso é necessário realizar os exames mencionados em primeiro lugar.
Embora as alterações cromossômicas da SD sejam comuns a todas as pessoas, nem todas apresentam as mesmas características, nem os mesmos traços físicos, tampouco as malformações. A única característica comum a todas as pessoas é o déficit intelectual. Não existem graus de SD; a variação das características e personalidades entre uma pessoa e outra é a mesma que existe entre as pessoas que não tem SD.
Cerca de 50% das crianças com SD apresentam problemas cardíacos, algumas vezes graves, necessitando de cirurgia nos primeiros anos de vida.
A intervenção médica pode acontecer com a finalidade principal de prevenção dos problemas de saúde que podem aparecer com maior freqüência na SD. Queremos destacar que a SD não é uma doença e sim uma alteração genética, que pode gerar problemas médicos associados.
Devemos olhar a pessoas com SD em sua singularidade, para que possa ter um pleno desenvolvimento enquanto sujeito.
O que é Síndrome de Turner?
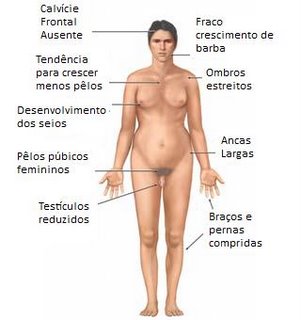 Sinônimos: Disgenesia gonadal, monossomia do X, Síndrome de Bonnevie-Ullrich
A síndrome de Turner é uma condição genética na qual uma mulher não tem o par normal de dois cromossomos X.
Sinônimos: Disgenesia gonadal, monossomia do X, Síndrome de Bonnevie-Ullrich
A síndrome de Turner é uma condição genética na qual uma mulher não tem o par normal de dois cromossomos X.
Sinônimos: Disgenesia gonadal, monossomia do X, Síndrome de Bonnevie-Ullrich
A síndrome de Turner é uma condição genética na qual uma mulher não tem o par normal de dois cromossomos X.
Causas
Os humanos possuem 46 cromossomos. Os cromossomos contêm todos os genes e o DNA, blocos de construção do corpo. Dois desses cromossomos, os cromossomos sexuais, determinam se será um menino ou uma menina. As mulheres normalmente possuem dois cromossomos do mesmo sexo, escrito como XX. Os homens têm um cromossomo X e um Y (escrito XY).
Na síndrome de Turner, as células carecem total ou parcialmente de um cromossomo X. Essa condição só ocorre em mulheres. Mais comumente, a paciente possui apenas um cromossomo X. Outras podem ter dois cromossomos X, mas um deles está incompleto. Às vezes, uma mulher tem algumas células com dois cromossomos X, mas outras células têm apenas um.
A síndrome de Turner ocorre em 1 a cada 2.000 partos de bebês vivos.
Os humanos possuem 46 cromossomos. Os cromossomos contêm todos os genes e o DNA, blocos de construção do corpo. Dois desses cromossomos, os cromossomos sexuais, determinam se será um menino ou uma menina. As mulheres normalmente possuem dois cromossomos do mesmo sexo, escrito como XX. Os homens têm um cromossomo X e um Y (escrito XY).
Na síndrome de Turner, as células carecem total ou parcialmente de um cromossomo X. Essa condição só ocorre em mulheres. Mais comumente, a paciente possui apenas um cromossomo X. Outras podem ter dois cromossomos X, mas um deles está incompleto. Às vezes, uma mulher tem algumas células com dois cromossomos X, mas outras células têm apenas um.
A síndrome de Turner ocorre em 1 a cada 2.000 partos de bebês vivos.
Exames
A Síndrome de Turner pode ser diagnosticada em qualquer fase da vida. Pode inclusive ser diagnosticada antes do nascimento, se a análise de cromossomos for feita nos exames de pré-natal.
O médico realizará um exame físico e buscará sinais de subdesenvolvimento. Bebês com síndrome de Turner têm frequentemente as mãos e os pés inchados.
Os seguintes exames podem ser realizados:
- Os níveis hormonais no sangue (hormônio luteinizante e hormônio folículo estimulante)
- Ecocardiograma
- Cariotipagem
- Ressonância magnética do tórax
- Ultrassonografia dos órgãos reprodutivos e rins
- Exame pélvico
A síndrome de Turner pode também alterar diversos níveis de estrogênio no sangue e na urina.
A Síndrome de Turner pode ser diagnosticada em qualquer fase da vida. Pode inclusive ser diagnosticada antes do nascimento, se a análise de cromossomos for feita nos exames de pré-natal.
O médico realizará um exame físico e buscará sinais de subdesenvolvimento. Bebês com síndrome de Turner têm frequentemente as mãos e os pés inchados.
Os seguintes exames podem ser realizados:
- Os níveis hormonais no sangue (hormônio luteinizante e hormônio folículo estimulante)
- Ecocardiograma
- Cariotipagem
- Ressonância magnética do tórax
- Ultrassonografia dos órgãos reprodutivos e rins
- Exame pélvico
A síndrome de Turner pode também alterar diversos níveis de estrogênio no sangue e na urina.
Síndrome de Klinefelter
 Sinônimos: Síndrome 47, XXY, Hipogonadismo
A síndrome de Klinefelter diz respeito à presença de um cromossomo X a mais no homem.
Sinônimos: Síndrome 47, XXY, Hipogonadismo
A síndrome de Klinefelter diz respeito à presença de um cromossomo X a mais no homem.
Sinônimos: Síndrome 47, XXY, Hipogonadismo
A síndrome de Klinefelter diz respeito à presença de um cromossomo X a mais no homem.
Causas
Os humanos possuem 46 cromossomos. Os cromossomos contêm todos os genes e o DNA, blocos de construção do corpo. Os dois cromossomos sexuais determinam se será um menino ou uma menina. As mulheres normalmente possuem dois cromossomos XX. Os homens normalmente possuem um X e um Y.
A síndrome de Klinefelter é um de vários problemas de cromossomos sexuais. A síndrome se dá em homens que têm pelo menos um cromossomo X extra. Normalmente, isso ocorre devido a um cromossomo X extra, que é escrito como XXY.
A síndrome de Klinefelter ocorre em 1 a cada 500 - 1.000 meninos recém-nascidos. Mulheres que engravidam depois dos 35 anos são um pouco mais propensas a terem um menino com a síndrome do que as mulheres mais jovens.
Os humanos possuem 46 cromossomos. Os cromossomos contêm todos os genes e o DNA, blocos de construção do corpo. Os dois cromossomos sexuais determinam se será um menino ou uma menina. As mulheres normalmente possuem dois cromossomos XX. Os homens normalmente possuem um X e um Y.
A síndrome de Klinefelter é um de vários problemas de cromossomos sexuais. A síndrome se dá em homens que têm pelo menos um cromossomo X extra. Normalmente, isso ocorre devido a um cromossomo X extra, que é escrito como XXY.
A síndrome de Klinefelter ocorre em 1 a cada 500 - 1.000 meninos recém-nascidos. Mulheres que engravidam depois dos 35 anos são um pouco mais propensas a terem um menino com a síndrome do que as mulheres mais jovens.
Exames
A Síndrome de Klinefelter pode ser diagnosticada primeiramente quando um homem procura o médico por causa de infertilidade. A infertilidade é o sintoma mais comum.
Os seguintes exames podem ser realizados:
- Cariotipagem
- Espermograma
Exames de sangue serão feitos para verificar os níveis hormonais, incluindo:
- Estradiol, um tipo de estrogênio
- Hormônio folículo estimulante
- Hormônio luteinizante
- Testosterona
SINDROME DE BURNOUT
O que é Burnout?
 O termo Burnout tem origem na língua inglesa, a partir da união de dois termos: burn e out, que respectivamente significam queimar e fora. A união dos termos é melhor traduzida por algo como “ser consumido pelo fogo”. A partir da década de 80, autores como Maslach passaram a usar esse termo para designar a síndrome decorrente da exaustão emocional humana, ou seja, uma condição em que o sujeito tem suas energias consumidas.A Síndrome de Burnout, como é chamada, compreende uma condição de estresse ligado ao trabalho, cuja definição ainda não é um conceito fechado. Alguns autores afirmam que a denominação deve levar em conta a questão da exaustão emocional, outros autores afirmam que essa síndrome é uma resposta inadequada do sujeito diante de uma situação de estresse crônico. Entre as principais características da exaustão característica da síndrome de Burnout, está a falta de energia, a sZensação de sobrecarga emocional constante e de esgotamento físico e mental.
Quais são os sintomas da Síndrome de Burnout?
A palavra síndrome designa um conjunto de sintomas, que podem ser físicos, psíquicos, de comportamento etc. No caso da Síndrome de Burnout, os sintomas mais expressivos são: crescimento da fadiga constante, distúrbios de sono, dores musculares, dores de cabeça e enxaquecas, problemas gastrointestinais, respiratórios, cardiovasculares. Em mulheres, as alterações no ciclo menstrual são um sintoma físico importante. Além desses, existem sintomas psicológicos como: dificuldade de concentração, lentificação ou alteração do pensamento, sentimentos negativos sobre o viver, trabalhar e ser, impaciência, irritabilidade, baixa autoestima, desconfiança, depressão, em alguns casos paranoia.
A partir desses sintomas, o sujeito acometido pela Síndrome de Burnout desenvolve comportamentos como: negligência ou perfeccionismo, agressividade nas relações cotidianas, perda da flexibilidade emocional e da capacidade de relaxar e planejar. Além disso, tende ao isolamento, à perda de interesse pelo trabalho e outras atividades.
Quais podem ser as causas?
As causas da Síndrome de Burnout compreendem um quadro multidimensional de fatores individuais e ambientais, que estão ligadas a uma percepção de desvalorização profissional. Isso significa dizer que não se pode reduzir a causa a fatores individuais como a personalidade ou algum tipo de propensão genética. O ambiente de trabalho e as condições de realização deste podem também determinar o adoecimento ou não do sujeito.
Alguns autores afirmam que a configuração do caso de Burnout passaria por estágios que vão desde uma necessidade de autoafirmação profissional, passando por estágios comuns de intensificação da dedicação ao trabalho que, levada a consequências extremas, resultaria no esgotamento característico da síndrome. Entre outros estágios, podemos destacar o caminho que passa pelo descaso crescente com relação às atividades de cuidado de si, como comer e dormir, acompanhado por um recalque de conflitos, caracterizado pelo não enfrentamento de situações que incomodam e pela negação dos problemas. Além desses, o sujeito passa por um processo de reinterpretação que faz com que coisas importantes sejam descartadas como inúteis.
Nesse quadro, já se pode falar em uma espécie de despersonalização, uma vez que o sujeito age de formas tão distintas que se torna “outra pessoa”, marcada por sinais de depressão, desesperança e exaustão, ou seja, uma espécie de colapso físico e mental que pode ser considerado quadro de emergência médica ou psicológica.
Quais são os tratamentos possíveis?
Como a grande maioria dos casos de adoecimento psicológico com consequências de somatização, o tratamento da Síndrome de Burnout deve compreender uma estratégia multidisciplinar: farmacológico, psicoterapêutico e médico. É sempre importante ressaltar a relevância de um diagnóstico realizado de maneira competente, para que não se cometam erros, como a confusão entre Burnout e Depressão, bastante comum nos estágios iniciais, pela similaridade de sintomas.
Com relação ao uso de medicamentos, o tratamento normalmente associa-se a antidepressivos e ansiolíticos. Este tratamento deve estar vinculado ao acompanhamento psicológico, que potencializa os efeitos do uso de medicamentos através da ressignificação e da retomada dos sentidos da história de vida do sujeito. Além desses, o acompanhamento médico e a alteração de hábitos são dimensões importantes. O encaminhamento para novas práticas cotidianas como exercícios físicos e de relaxamento é de extrema importância.
O termo Burnout tem origem na língua inglesa, a partir da união de dois termos: burn e out, que respectivamente significam queimar e fora. A união dos termos é melhor traduzida por algo como “ser consumido pelo fogo”. A partir da década de 80, autores como Maslach passaram a usar esse termo para designar a síndrome decorrente da exaustão emocional humana, ou seja, uma condição em que o sujeito tem suas energias consumidas.A Síndrome de Burnout, como é chamada, compreende uma condição de estresse ligado ao trabalho, cuja definição ainda não é um conceito fechado. Alguns autores afirmam que a denominação deve levar em conta a questão da exaustão emocional, outros autores afirmam que essa síndrome é uma resposta inadequada do sujeito diante de uma situação de estresse crônico. Entre as principais características da exaustão característica da síndrome de Burnout, está a falta de energia, a sZensação de sobrecarga emocional constante e de esgotamento físico e mental.
Quais são os sintomas da Síndrome de Burnout?
A palavra síndrome designa um conjunto de sintomas, que podem ser físicos, psíquicos, de comportamento etc. No caso da Síndrome de Burnout, os sintomas mais expressivos são: crescimento da fadiga constante, distúrbios de sono, dores musculares, dores de cabeça e enxaquecas, problemas gastrointestinais, respiratórios, cardiovasculares. Em mulheres, as alterações no ciclo menstrual são um sintoma físico importante. Além desses, existem sintomas psicológicos como: dificuldade de concentração, lentificação ou alteração do pensamento, sentimentos negativos sobre o viver, trabalhar e ser, impaciência, irritabilidade, baixa autoestima, desconfiança, depressão, em alguns casos paranoia.
A partir desses sintomas, o sujeito acometido pela Síndrome de Burnout desenvolve comportamentos como: negligência ou perfeccionismo, agressividade nas relações cotidianas, perda da flexibilidade emocional e da capacidade de relaxar e planejar. Além disso, tende ao isolamento, à perda de interesse pelo trabalho e outras atividades.
Quais podem ser as causas?
As causas da Síndrome de Burnout compreendem um quadro multidimensional de fatores individuais e ambientais, que estão ligadas a uma percepção de desvalorização profissional. Isso significa dizer que não se pode reduzir a causa a fatores individuais como a personalidade ou algum tipo de propensão genética. O ambiente de trabalho e as condições de realização deste podem também determinar o adoecimento ou não do sujeito.
Alguns autores afirmam que a configuração do caso de Burnout passaria por estágios que vão desde uma necessidade de autoafirmação profissional, passando por estágios comuns de intensificação da dedicação ao trabalho que, levada a consequências extremas, resultaria no esgotamento característico da síndrome. Entre outros estágios, podemos destacar o caminho que passa pelo descaso crescente com relação às atividades de cuidado de si, como comer e dormir, acompanhado por um recalque de conflitos, caracterizado pelo não enfrentamento de situações que incomodam e pela negação dos problemas. Além desses, o sujeito passa por um processo de reinterpretação que faz com que coisas importantes sejam descartadas como inúteis.
Nesse quadro, já se pode falar em uma espécie de despersonalização, uma vez que o sujeito age de formas tão distintas que se torna “outra pessoa”, marcada por sinais de depressão, desesperança e exaustão, ou seja, uma espécie de colapso físico e mental que pode ser considerado quadro de emergência médica ou psicológica.
Quais são os tratamentos possíveis?
Como a grande maioria dos casos de adoecimento psicológico com consequências de somatização, o tratamento da Síndrome de Burnout deve compreender uma estratégia multidisciplinar: farmacológico, psicoterapêutico e médico. É sempre importante ressaltar a relevância de um diagnóstico realizado de maneira competente, para que não se cometam erros, como a confusão entre Burnout e Depressão, bastante comum nos estágios iniciais, pela similaridade de sintomas.
Com relação ao uso de medicamentos, o tratamento normalmente associa-se a antidepressivos e ansiolíticos. Este tratamento deve estar vinculado ao acompanhamento psicológico, que potencializa os efeitos do uso de medicamentos através da ressignificação e da retomada dos sentidos da história de vida do sujeito. Além desses, o acompanhamento médico e a alteração de hábitos são dimensões importantes. O encaminhamento para novas práticas cotidianas como exercícios físicos e de relaxamento é de extrema importância.
SINDROME DE EDWARDS
A síndrome de Edwards, também conhecida como trissomia 18, é uma síndrome genética causada por uma trissomia do cromossomo 18.
 Foi descrita primeiramente em 1960, por John H. Edwards, em recém-nascidos que apresentavam malformações congênitas múltiplas e retardamento mental. Esta foi a segunda síndrome revelada no homem, sendo que a primeira foi a síndrome de Down ou trissomia 21.
Foi descrita primeiramente em 1960, por John H. Edwards, em recém-nascidos que apresentavam malformações congênitas múltiplas e retardamento mental. Esta foi a segunda síndrome revelada no homem, sendo que a primeira foi a síndrome de Down ou trissomia 21.
Acomete 1 em cada 8.000 nascidos, sendo o sexo feminino mais comumente afetado. Entretanto, acredita-se que 95% dos casos dessa síndrome resultem em aborto espontâneo durante a gestação. A expectativa de vida para um portador da síndrome de Edwards é baixa; todavia, já foram descritos casos de adolescentes com 15 anos de idade portadores da afecção.
A maior parte dos pacientes portadores dessa síndrome apresenta trissomia regular sem mosaicismo, ou seja, cariótipo47, XX ou XY, +18. Dentre os restantes, aproximadamente metade é formada por casos de mosaicismo e outra parcela por problemas mais complexos, como aneuploidias duplas, translocações. Destes, cerca de 80% dos casos são resultantes de uma translocação abrangendo todo ou quase todo o cromossomo 18, sendo que este pode ser recebido ou adquirido novamente a partir de um progenitor transportador.
As características apresentadas pelos portadores da trissomia 18 são retardamento físico, choro fraco, hipotonia seguida de hipertonia, hipoplasia da musculatura esquelética e do tecido adiposo subcutâneo, redução de resposta a estímulos sonoros, retardo mental e diversas características físicas, como:
- Crânio disfórmico;
- Face triangular com testa alta e plana;
- Maxilares recuados;
Orelhas mal formadas e baixas; - Occipital proeminente;
- Lábio leporino e/ou fenda palatina;
- Pescoço curto com pêlos em excesso;
- Externo curto;
- Mamilos pequenos;
- Presença de hérnia inguinal ou umbilical;
- Manutenção dos punhos cerrados é característico;
- Pé torto congênito é comum;
- Encurtamento do hálux (dedão do pé);
- Rugas nas palmas das mãos e plantas dos pés;
- Nos meninos é comum a ocorrência de criptorquidia, já nas meninas é comum a hipertrofia de clitóris com hipoplasia dos grandes lábios;
Diversas malformações congênitas podem ser encontradas, afetando o cérebro, coração, rins e aparelho gastrointestinal. Entre as malformações cardíacas mais frequentes, que normalmente é a causa do óbito nesses pacientes, está a comunicação interventricular e a persistência do ducto arterial. Também observa-se com frequência a presença de tecido pancreático heterotrópico, eventração diafragmática, divertículo de Meckel e diferentes tipos de displasias renais.
Ainda dentro da barriga, já é possível detectar e presença de anomalias nos fetos. O exame ultra-sonográfico transvaginal, entre 10 a 14 semanas de gestação, possibilita estimar a espessura do “espaço escuro” existente entre a pele e o tecido subcutâneo, que reveste a coluna cervical fetal, detectando, deste modo, alterações no feto.
O diagnóstico diferencial deve ser feito com a síndrome da trissomia 13 (ou síndrome de Patau), pois em ambas os indivíduos podem apresentar lábio leporino e/ou fenda palatina.
Quando há o aparecimento dessa síndrome, aconselha-se procurar aconselhamento genético, para que seja realizado um estudo genético.
O prognóstico para indivíduos que nascem com essa doença genética é ruim, sendo a sobrevida da maioria desses pacientes é de 2 a 3 meses para os meninos e 10 meses para as meninas, muito dificilmente ultrapassando os 2 anos de vida; os pacientes que possuem o mosaicismo podem sobreviver por mais tempo.
SINDROME DE RETT
 A Síndrome de Rett é definida como uma desordem do desenvolvimento neurológico relativamente rara, tendo sido reconhecida pelo mundo no início da década de 1980. Desde então, diversos estudos já apontaram que pode ocorrer em qualquer grupo étnico com aproximadamente a mesma incidência. A prevalência da Síndrome de Rett é de uma em cada 10.000-20.000 pessoas do sexo feminino.
A Síndrome de Rett é definida como uma desordem do desenvolvimento neurológico relativamente rara, tendo sido reconhecida pelo mundo no início da década de 1980. Desde então, diversos estudos já apontaram que pode ocorrer em qualquer grupo étnico com aproximadamente a mesma incidência. A prevalência da Síndrome de Rett é de uma em cada 10.000-20.000 pessoas do sexo feminino.
Desde que foi identificada, sempre foi vislumbrada a natureza genética dessa desordem, primeiro por afetar predominantemente o sexo feminino, e também pelos raros casos familiares, embora se trate de síndrome de ocorrência esporádica em 95,5% dos casos, e o risco de casos familiares seja inferior a 0,5%. Desde 1999, já se sabe que a grande maioria das meninas e mulheres que preenchem os critérios para a Síndrome de Rett apresenta mutações no gene MECP2.
Durante os últimos 25 anos, os conhecimentos sobre as características clínicas e a história natural da Síndrome de Rett evoluíram de maneira surpreendente. Entretanto, ainda se trata de condição muito desconhecida para segmentos sociais e científicos importantes: ainda há muitos médicos, terapeutas e educadores que não fazem ideia do que seja a Síndrome de Rett, e muitos dos que já ouviram falar sobre ela permanecem relativamente desinformados sobre os avanços no conhecimento clínico e terapêutico adquiridos especialmente nesta última década.
Causa
A Síndrome de Rett é uma doença de causa genética e está associada a mutações no gene MECP2 (do inglês, methyl-CpG-binding protein 2), localizado no cromossomo X.
O MECP2 é de extrema importância no controle de outros genes, e a proteína por ele codificada (também chamada de MeCP2) atua como que “desligando” determinados genes durante fases específicas do desenvolvimento neuronal.
A base molecular da Síndrome de Rett consiste no fato de o gene MECP2 mutado codificar uma proteína defeituosa, incapaz de exercer adequadamente sua função biológica, o que faz com que os genes que deveriam estar silenciados (“desligados”) durante determinadas fases do desenvolvimento dos neurônios permaneçam ativos (“ligados”), resultando em prejuízos ao desenvolvimento do sistema nervoso central.
Aproximadamente 90% dos casos de Síndrome de Rett são devidos a quatro mutações do gene MECP2 que resultam em manifestações clínicas menos graves (R106W, R133C, T158M, R306C) e a outras quatro deleções amplas que causam uma destruição mais grave do gene e, consequentemente, resultam em manifestações clínicas mais severas (R168X, R255X, R270X, R294X). Nos 10% restantes, acredita-se que as mutações estejam localizadas em outras regiões do gene ou em outros genes associados à doença, como o gene CDKL5 (do inglês, cyclin-dependent kinase-like 5), também localizado no cromossomo X, e no qual mutações foram detectadas em pacientes com SR, e talvez em outros genes que ainda não foram relacionados à doença.
Cerca de 99,5% dos casos de Síndrome de Rett são esporádicos, ou seja, sem antecedentes familiares. A análise da origem parental (dos pais) nos casos esporádicos revelou que, na maioria deles, as mutações ocorreram de novo no alelo de origem paterna. Isso explicaria a predominância de Síndrome de Rett clássica em meninas, já que os meninos não herdam o cromossomo X paterno, mas sim o materno.
Por outro lado, nos raros casos familiais (somente 0,5%), o alelo mutado é proveniente da mãe, que não apresenta o quadro clínico de Síndrome de Rett. Esses casos poderiam ser explicados: (1) pela inativação favorável do cromossomo X da mãe que tem a mutação; ou (2) pela ocorrência de mosaicismo gonadal, quando o alelo mutado é encontrado apenas nas células da linhagem germinativa materna.
Através da análise molecular do gene MECP2 da paciente e de sua mãe, é possível verificar se a mutação foi herdada da mãe, para, assim, procurar aconselhamento genético. Embora os casos familiais sejam raros, é indicado avaliar a possibilidade de recorrência da doença na família, inclusive utilizando-se o diagnóstico pré-natal, que envolve a coleta de material biológico do feto pela amniocentese ou pela punção de vilosidades coriônicas e pesquisa de mutações no MECP2 nesse material.
SINDROME DE PATAU
 A síndrome de Patau é uma anomalia cromossômica resultante da trissomia do cromossomo 13.
Foi descrita primeiramente por Klaus Patau, no ano de 1960. Este, por sua vez, observou casos de malformações múltiplas em neonato, que apresentavam trissomia do cromossomo 13.
Esta síndrome também recebe o nome de síndrome Bartholin-Patau, uma vez que Thomas Bartholin, um dinamarquês, descreveu anteriormente à Patau, no ano de 1956, o quadro clínico de crianças que apresentavam tal deficiência.
Normalmente, os seres humanos apresentam 23 pares de cromossomos, ou seja, são 46 cromossomos repartidos em 23 pares de 2 cromossomos. A trissomia ocorre quando um indivíduo apresenta 3 cromossomos no grupo 13.
A síndrome de Patau é uma anomalia cromossômica resultante da trissomia do cromossomo 13.
Foi descrita primeiramente por Klaus Patau, no ano de 1960. Este, por sua vez, observou casos de malformações múltiplas em neonato, que apresentavam trissomia do cromossomo 13.
Esta síndrome também recebe o nome de síndrome Bartholin-Patau, uma vez que Thomas Bartholin, um dinamarquês, descreveu anteriormente à Patau, no ano de 1956, o quadro clínico de crianças que apresentavam tal deficiência.
Normalmente, os seres humanos apresentam 23 pares de cromossomos, ou seja, são 46 cromossomos repartidos em 23 pares de 2 cromossomos. A trissomia ocorre quando um indivíduo apresenta 3 cromossomos no grupo 13.
Esta anomalia origina-se no gameta feminino (óvulo) e estudos revelam que entre 40 a 60% das crianças que apresentam esta síndrome são proles de mães com idade superior a 35 anos. A não disjunção dos cromossomos durante o processo de anáfase 1 da meiose, origina gametas com 24 cromátides, ou seja, o gameta apresenta um par de cromossomos 13 que, em conjunto com o cromossomo 13 do espermatozóide, resulta em um embrião com trissomia.
Os portadores da trissomia 13 apresentam graves malformações do sistema nervoso central, como, por exemplo, arrinencefalia; baixo peso ao nascimento; defeitos na formação dos olhos ou ausência dos mesmos; problemas auditivos; anormalidades no controle da respiração; fenda palatina e/ou lábio leporino; rins policísticos; malformação das mãos. Defeitos cardíacos congênitos também estão comumente presente nesta síndrome, bem como defeitos urogenitais que englobam criptorquidia nos meninos, útero bicornado e ovários hipoplásicos nas meninas. Tanto nas mãos quanto nos pés pode haver polidactilia.
Hoje em dia, já estão disponíveis diferentes exames capazes de identificar com detalhes os cromossomos afetados e seus segmentos, alcançando-se um diagnóstico preciso da síndrome antes mesmo do nascimento. Após detectada a trissomia, é necessária a realização de alguns exames complementares para confirmação do diagnóstico.
A incidência da trissomia 13 é muito mais elevada em crianças do sexo feminino do que no masculino, afetando em torno de 1 em cada 7.000 nascidos vivos. Todavia, acredita-se que somente 2,5% dos fetos com trissomia 13 nasçam vivos, sendo esta uma das principais causas de aborto espontâneo nos três primeiros meses de gestação.
Por esta ser uma moléstia grave e que apresenta diferentes malformações congênitas, o prognóstico de sobrevivência é muito curto, sendo que a maior parte dos nascidos vivos morre dentro do primeiro mês de vida. No entanto, há parcos relatos de crianças portadoras da síndrome que sobreviveu até os 10 anos de idade.
FONTES
http://pt.wikipedia.org/wiki/Síndrome_de_Edwards
A Síndrome de Klinefelter pode ser diagnosticada primeiramente quando um homem procura o médico por causa de infertilidade. A infertilidade é o sintoma mais comum.
Os seguintes exames podem ser realizados:
- Cariotipagem
- Espermograma
Exames de sangue serão feitos para verificar os níveis hormonais, incluindo:
- Estradiol, um tipo de estrogênio
- Hormônio folículo estimulante
- Hormônio luteinizante
- Testosterona
SINDROME DE BURNOUT
O que é Burnout?
O termo Burnout tem origem na língua inglesa, a partir da união de dois termos: burn e out, que respectivamente significam queimar e fora. A união dos termos é melhor traduzida por algo como “ser consumido pelo fogo”. A partir da década de 80, autores como Maslach passaram a usar esse termo para designar a síndrome decorrente da exaustão emocional humana, ou seja, uma condição em que o sujeito tem suas energias consumidas.A Síndrome de Burnout, como é chamada, compreende uma condição de estresse ligado ao trabalho, cuja definição ainda não é um conceito fechado. Alguns autores afirmam que a denominação deve levar em conta a questão da exaustão emocional, outros autores afirmam que essa síndrome é uma resposta inadequada do sujeito diante de uma situação de estresse crônico. Entre as principais características da exaustão característica da síndrome de Burnout, está a falta de energia, a sZensação de sobrecarga emocional constante e de esgotamento físico e mental.
Quais são os sintomas da Síndrome de Burnout?
A palavra síndrome designa um conjunto de sintomas, que podem ser físicos, psíquicos, de comportamento etc. No caso da Síndrome de Burnout, os sintomas mais expressivos são: crescimento da fadiga constante, distúrbios de sono, dores musculares, dores de cabeça e enxaquecas, problemas gastrointestinais, respiratórios, cardiovasculares. Em mulheres, as alterações no ciclo menstrual são um sintoma físico importante. Além desses, existem sintomas psicológicos como: dificuldade de concentração, lentificação ou alteração do pensamento, sentimentos negativos sobre o viver, trabalhar e ser, impaciência, irritabilidade, baixa autoestima, desconfiança, depressão, em alguns casos paranoia.
A partir desses sintomas, o sujeito acometido pela Síndrome de Burnout desenvolve comportamentos como: negligência ou perfeccionismo, agressividade nas relações cotidianas, perda da flexibilidade emocional e da capacidade de relaxar e planejar. Além disso, tende ao isolamento, à perda de interesse pelo trabalho e outras atividades.
Quais podem ser as causas?
As causas da Síndrome de Burnout compreendem um quadro multidimensional de fatores individuais e ambientais, que estão ligadas a uma percepção de desvalorização profissional. Isso significa dizer que não se pode reduzir a causa a fatores individuais como a personalidade ou algum tipo de propensão genética. O ambiente de trabalho e as condições de realização deste podem também determinar o adoecimento ou não do sujeito.
Alguns autores afirmam que a configuração do caso de Burnout passaria por estágios que vão desde uma necessidade de autoafirmação profissional, passando por estágios comuns de intensificação da dedicação ao trabalho que, levada a consequências extremas, resultaria no esgotamento característico da síndrome. Entre outros estágios, podemos destacar o caminho que passa pelo descaso crescente com relação às atividades de cuidado de si, como comer e dormir, acompanhado por um recalque de conflitos, caracterizado pelo não enfrentamento de situações que incomodam e pela negação dos problemas. Além desses, o sujeito passa por um processo de reinterpretação que faz com que coisas importantes sejam descartadas como inúteis.
Nesse quadro, já se pode falar em uma espécie de despersonalização, uma vez que o sujeito age de formas tão distintas que se torna “outra pessoa”, marcada por sinais de depressão, desesperança e exaustão, ou seja, uma espécie de colapso físico e mental que pode ser considerado quadro de emergência médica ou psicológica.
Quais são os tratamentos possíveis?
Como a grande maioria dos casos de adoecimento psicológico com consequências de somatização, o tratamento da Síndrome de Burnout deve compreender uma estratégia multidisciplinar: farmacológico, psicoterapêutico e médico. É sempre importante ressaltar a relevância de um diagnóstico realizado de maneira competente, para que não se cometam erros, como a confusão entre Burnout e Depressão, bastante comum nos estágios iniciais, pela similaridade de sintomas.
Com relação ao uso de medicamentos, o tratamento normalmente associa-se a antidepressivos e ansiolíticos. Este tratamento deve estar vinculado ao acompanhamento psicológico, que potencializa os efeitos do uso de medicamentos através da ressignificação e da retomada dos sentidos da história de vida do sujeito. Além desses, o acompanhamento médico e a alteração de hábitos são dimensões importantes. O encaminhamento para novas práticas cotidianas como exercícios físicos e de relaxamento é de extrema importância.
SINDROME DE EDWARDS
A síndrome de Edwards, também conhecida como trissomia 18, é uma síndrome genética causada por uma trissomia do cromossomo 18.
 Foi descrita primeiramente em 1960, por John H. Edwards, em recém-nascidos que apresentavam malformações congênitas múltiplas e retardamento mental. Esta foi a segunda síndrome revelada no homem, sendo que a primeira foi a síndrome de Down ou trissomia 21.
Foi descrita primeiramente em 1960, por John H. Edwards, em recém-nascidos que apresentavam malformações congênitas múltiplas e retardamento mental. Esta foi a segunda síndrome revelada no homem, sendo que a primeira foi a síndrome de Down ou trissomia 21.
A maior parte dos pacientes portadores dessa síndrome apresenta trissomia regular sem mosaicismo, ou seja, cariótipo47, XX ou XY, +18. Dentre os restantes, aproximadamente metade é formada por casos de mosaicismo e outra parcela por problemas mais complexos, como aneuploidias duplas, translocações. Destes, cerca de 80% dos casos são resultantes de uma translocação abrangendo todo ou quase todo o cromossomo 18, sendo que este pode ser recebido ou adquirido novamente a partir de um progenitor transportador.
As características apresentadas pelos portadores da trissomia 18 são retardamento físico, choro fraco, hipotonia seguida de hipertonia, hipoplasia da musculatura esquelética e do tecido adiposo subcutâneo, redução de resposta a estímulos sonoros, retardo mental e diversas características físicas, como:
- Crânio disfórmico;
- Face triangular com testa alta e plana;
- Maxilares recuados;
Orelhas mal formadas e baixas; - Occipital proeminente;
- Lábio leporino e/ou fenda palatina;
- Pescoço curto com pêlos em excesso;
- Externo curto;
- Mamilos pequenos;
- Presença de hérnia inguinal ou umbilical;
- Manutenção dos punhos cerrados é característico;
- Pé torto congênito é comum;
- Encurtamento do hálux (dedão do pé);
- Rugas nas palmas das mãos e plantas dos pés;
- Nos meninos é comum a ocorrência de criptorquidia, já nas meninas é comum a hipertrofia de clitóris com hipoplasia dos grandes lábios;
Ainda dentro da barriga, já é possível detectar e presença de anomalias nos fetos. O exame ultra-sonográfico transvaginal, entre 10 a 14 semanas de gestação, possibilita estimar a espessura do “espaço escuro” existente entre a pele e o tecido subcutâneo, que reveste a coluna cervical fetal, detectando, deste modo, alterações no feto.
O diagnóstico diferencial deve ser feito com a síndrome da trissomia 13 (ou síndrome de Patau), pois em ambas os indivíduos podem apresentar lábio leporino e/ou fenda palatina.
Quando há o aparecimento dessa síndrome, aconselha-se procurar aconselhamento genético, para que seja realizado um estudo genético.
O prognóstico para indivíduos que nascem com essa doença genética é ruim, sendo a sobrevida da maioria desses pacientes é de 2 a 3 meses para os meninos e 10 meses para as meninas, muito dificilmente ultrapassando os 2 anos de vida; os pacientes que possuem o mosaicismo podem sobreviver por mais tempo.
SINDROME DE RETT
A Síndrome de Rett é definida como uma desordem do desenvolvimento neurológico relativamente rara, tendo sido reconhecida pelo mundo no início da década de 1980. Desde então, diversos estudos já apontaram que pode ocorrer em qualquer grupo étnico com aproximadamente a mesma incidência. A prevalência da Síndrome de Rett é de uma em cada 10.000-20.000 pessoas do sexo feminino.Desde que foi identificada, sempre foi vislumbrada a natureza genética dessa desordem, primeiro por afetar predominantemente o sexo feminino, e também pelos raros casos familiares, embora se trate de síndrome de ocorrência esporádica em 95,5% dos casos, e o risco de casos familiares seja inferior a 0,5%. Desde 1999, já se sabe que a grande maioria das meninas e mulheres que preenchem os critérios para a Síndrome de Rett apresenta mutações no gene MECP2.
Durante os últimos 25 anos, os conhecimentos sobre as características clínicas e a história natural da Síndrome de Rett evoluíram de maneira surpreendente. Entretanto, ainda se trata de condição muito desconhecida para segmentos sociais e científicos importantes: ainda há muitos médicos, terapeutas e educadores que não fazem ideia do que seja a Síndrome de Rett, e muitos dos que já ouviram falar sobre ela permanecem relativamente desinformados sobre os avanços no conhecimento clínico e terapêutico adquiridos especialmente nesta última década.
| Causa | |||
A Síndrome de Rett é uma doença de causa genética e está associada a mutações no gene MECP2 (do inglês, methyl-CpG-binding protein 2), localizado no cromossomo X.
O MECP2 é de extrema importância no controle de outros genes, e a proteína por ele codificada (também chamada de MeCP2) atua como que “desligando” determinados genes durante fases específicas do desenvolvimento neuronal.
A base molecular da Síndrome de Rett consiste no fato de o gene MECP2 mutado codificar uma proteína defeituosa, incapaz de exercer adequadamente sua função biológica, o que faz com que os genes que deveriam estar silenciados (“desligados”) durante determinadas fases do desenvolvimento dos neurônios permaneçam ativos (“ligados”), resultando em prejuízos ao desenvolvimento do sistema nervoso central.
Aproximadamente 90% dos casos de Síndrome de Rett são devidos a quatro mutações do gene MECP2 que resultam em manifestações clínicas menos graves (R106W, R133C, T158M, R306C) e a outras quatro deleções amplas que causam uma destruição mais grave do gene e, consequentemente, resultam em manifestações clínicas mais severas (R168X, R255X, R270X, R294X). Nos 10% restantes, acredita-se que as mutações estejam localizadas em outras regiões do gene ou em outros genes associados à doença, como o gene CDKL5 (do inglês, cyclin-dependent kinase-like 5), também localizado no cromossomo X, e no qual mutações foram detectadas em pacientes com SR, e talvez em outros genes que ainda não foram relacionados à doença.
Cerca de 99,5% dos casos de Síndrome de Rett são esporádicos, ou seja, sem antecedentes familiares. A análise da origem parental (dos pais) nos casos esporádicos revelou que, na maioria deles, as mutações ocorreram de novo no alelo de origem paterna. Isso explicaria a predominância de Síndrome de Rett clássica em meninas, já que os meninos não herdam o cromossomo X paterno, mas sim o materno.
Por outro lado, nos raros casos familiais (somente 0,5%), o alelo mutado é proveniente da mãe, que não apresenta o quadro clínico de Síndrome de Rett. Esses casos poderiam ser explicados: (1) pela inativação favorável do cromossomo X da mãe que tem a mutação; ou (2) pela ocorrência de mosaicismo gonadal, quando o alelo mutado é encontrado apenas nas células da linhagem germinativa materna.
Através da análise molecular do gene MECP2 da paciente e de sua mãe, é possível verificar se a mutação foi herdada da mãe, para, assim, procurar aconselhamento genético. Embora os casos familiais sejam raros, é indicado avaliar a possibilidade de recorrência da doença na família, inclusive utilizando-se o diagnóstico pré-natal, que envolve a coleta de material biológico do feto pela amniocentese ou pela punção de vilosidades coriônicas e pesquisa de mutações no MECP2 nesse material.
O MECP2 é de extrema importância no controle de outros genes, e a proteína por ele codificada (também chamada de MeCP2) atua como que “desligando” determinados genes durante fases específicas do desenvolvimento neuronal.
A base molecular da Síndrome de Rett consiste no fato de o gene MECP2 mutado codificar uma proteína defeituosa, incapaz de exercer adequadamente sua função biológica, o que faz com que os genes que deveriam estar silenciados (“desligados”) durante determinadas fases do desenvolvimento dos neurônios permaneçam ativos (“ligados”), resultando em prejuízos ao desenvolvimento do sistema nervoso central.
Aproximadamente 90% dos casos de Síndrome de Rett são devidos a quatro mutações do gene MECP2 que resultam em manifestações clínicas menos graves (R106W, R133C, T158M, R306C) e a outras quatro deleções amplas que causam uma destruição mais grave do gene e, consequentemente, resultam em manifestações clínicas mais severas (R168X, R255X, R270X, R294X). Nos 10% restantes, acredita-se que as mutações estejam localizadas em outras regiões do gene ou em outros genes associados à doença, como o gene CDKL5 (do inglês, cyclin-dependent kinase-like 5), também localizado no cromossomo X, e no qual mutações foram detectadas em pacientes com SR, e talvez em outros genes que ainda não foram relacionados à doença.
Cerca de 99,5% dos casos de Síndrome de Rett são esporádicos, ou seja, sem antecedentes familiares. A análise da origem parental (dos pais) nos casos esporádicos revelou que, na maioria deles, as mutações ocorreram de novo no alelo de origem paterna. Isso explicaria a predominância de Síndrome de Rett clássica em meninas, já que os meninos não herdam o cromossomo X paterno, mas sim o materno.
Por outro lado, nos raros casos familiais (somente 0,5%), o alelo mutado é proveniente da mãe, que não apresenta o quadro clínico de Síndrome de Rett. Esses casos poderiam ser explicados: (1) pela inativação favorável do cromossomo X da mãe que tem a mutação; ou (2) pela ocorrência de mosaicismo gonadal, quando o alelo mutado é encontrado apenas nas células da linhagem germinativa materna.
Através da análise molecular do gene MECP2 da paciente e de sua mãe, é possível verificar se a mutação foi herdada da mãe, para, assim, procurar aconselhamento genético. Embora os casos familiais sejam raros, é indicado avaliar a possibilidade de recorrência da doença na família, inclusive utilizando-se o diagnóstico pré-natal, que envolve a coleta de material biológico do feto pela amniocentese ou pela punção de vilosidades coriônicas e pesquisa de mutações no MECP2 nesse material.
SINDROME DE PATAU
 A síndrome de Patau é uma anomalia cromossômica resultante da trissomia do cromossomo 13.
A síndrome de Patau é uma anomalia cromossômica resultante da trissomia do cromossomo 13.
Foi descrita primeiramente por Klaus Patau, no ano de 1960. Este, por sua vez, observou casos de malformações múltiplas em neonato, que apresentavam trissomia do cromossomo 13.
Esta síndrome também recebe o nome de síndrome Bartholin-Patau, uma vez que Thomas Bartholin, um dinamarquês, descreveu anteriormente à Patau, no ano de 1956, o quadro clínico de crianças que apresentavam tal deficiência.
Normalmente, os seres humanos apresentam 23 pares de cromossomos, ou seja, são 46 cromossomos repartidos em 23 pares de 2 cromossomos. A trissomia ocorre quando um indivíduo apresenta 3 cromossomos no grupo 13.
Os portadores da trissomia 13 apresentam graves malformações do sistema nervoso central, como, por exemplo, arrinencefalia; baixo peso ao nascimento; defeitos na formação dos olhos ou ausência dos mesmos; problemas auditivos; anormalidades no controle da respiração; fenda palatina e/ou lábio leporino; rins policísticos; malformação das mãos. Defeitos cardíacos congênitos também estão comumente presente nesta síndrome, bem como defeitos urogenitais que englobam criptorquidia nos meninos, útero bicornado e ovários hipoplásicos nas meninas. Tanto nas mãos quanto nos pés pode haver polidactilia.
A incidência da trissomia 13 é muito mais elevada em crianças do sexo feminino do que no masculino, afetando em torno de 1 em cada 7.000 nascidos vivos. Todavia, acredita-se que somente 2,5% dos fetos com trissomia 13 nasçam vivos, sendo esta uma das principais causas de aborto espontâneo nos três primeiros meses de gestação.
Por esta ser uma moléstia grave e que apresenta diferentes malformações congênitas, o prognóstico de sobrevivência é muito curto, sendo que a maior parte dos nascidos vivos morre dentro do primeiro mês de vida. No entanto, há parcos relatos de crianças portadoras da síndrome que sobreviveu até os 10 anos de idade.
Nenhum comentário :
Postar um comentário